
Our knowledge of the human genome has increased exponentially since British chemist Frederick Sanger won the 1980 Nobel Prize for his eponymous approach to DNA sequencing. While “Sanger sequencing” offered great accuracy, it was also restricted to a single DNA fragment. As the Human Genome Project demonstrated, Sanger sequencing offered low throughput, but very high cost--the project took more than a decade and cost more than $3 billion.
The mid-2000’s brought next generation sequencing (NGS), a technique that offers much greater speed at a much lower cost; by 2011, an entire human genome could be sequenced in a few weeks, for about $10,000. NGS has proven revolutionary in the study of genetic diseases because it allows genome-wide detection of variants, including novel ones. NGS can also be applied to pharmacogenetic problems, allowing researchers to evaluate not only why certain drugs are ineffective for specific patients, but also to predict the success of a drug based on genetic factors.
According to the National Human Genome Research Institute, the next phase of genetic research is characterized by cooperative, large-scale initiatives like the Encyclopedia of DNA Elements (ENCODE) Consortium. These initiatives require a much more team-oriented approach to research because they bring together researchers and institutions from all over the world.
The collaborative nature of human genome research makes it an ideal candidate for cloud computing. Furthermore, NGS also presents a significant challenge in terms of data storage and processing, which can also be addressed through cloud technologies. The following case studies included in UberCloud's Compendium of Life Sciences, explore applications and benefits of cloud technologies for human genome research.
Case Study: NGS Data Analysis in the Cloud
NGS is a high-throughput sequencing technology that is revolutionizing medicine; its accuracy and breadth of coverage make it useful for diagnosing and treating hereditary diseases and cancer, as well as infectious disease. However, the advantages of NGS lead to corollary challenges: storing, sharing, and analyzing the data strains disk space, network bandwidth and processor resources.
Cloud computing can alleviate some of these problems by flexibly providing virtualized compute and storage resources on-demand. Cluster computing, running on top of virtualized infrastructure can assist in coordinating the compute and storage resources for high-throughput applications, such as when processing NGS data. Bioinformatics workflow services such as those provided by Nimbus Informatics are built on top of infrastructure providers such as Amazon Web Services (AWS) and promise to provide an end-to-end platform that automates storing, organizing and processing large amounts of data in the cloud.
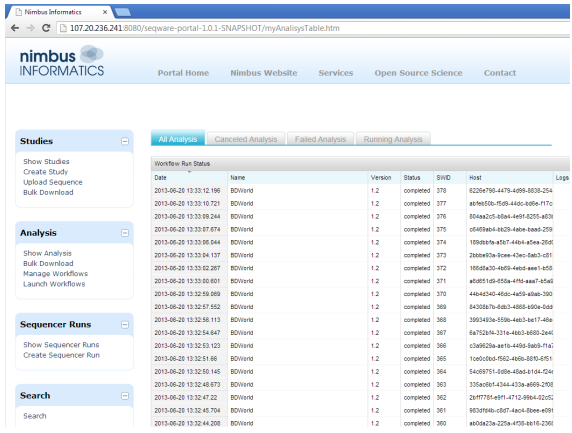 |
| Workflow run status is displayed in a table. The team ran approximately 30 workflows simultaneously on Nimbus' AWS-based system. |
In this case study, the research team used SeqWare AMI, a comprehensive, open-source toolbox billed as a “genome center in a box.” SeqWare AMI is pre-set to run workflows directly on the EC2 instance, without an external cluster, making it a great option for development and testing. To test the scalability of the system by processing the samples in parallel, the team used Nimbus Informatics, a provider of hosted computational workflows that enable the analysis of complex genomics data for customers lacking a local HPC infrastructure.
The team identified three benefits of this solution:
- By partnering with Nimbus, the end user can write workflows using an open-source infrastructure, increasing transparency.
- Because of the open nature of the software, end users can create their own analytical workflows using standard tools, but customize and test them locally, with customization for their particular analytical challenges.
- The cloud-based hosting of the SeqWare system allowed users to build and run their own SeqWare infrastructure on their own hardware. This meant that users could write and test their workflows locally, upload them to the Nimbus infrastructure and simply “order” workflows or specified samples.
Case Study: Using Cloud Computing to Perform Molecular Dynamics Simulations of the Mutant PI3Kα Protein
 |
| The figure above illustrates the solvated PI3Kα in a water box. |
Cancer is a leading cause of death worldwide. The World Health Organization estimates that by 2030, 21 million people will die from the disease each year. One of the signaling pathways which, when deregulated, becomes centrally involved in several types of cancers, like colon, mammary, and endometrial tumorigenesis, is served by phosphoinositide-3-kinase alpha (PI3Kα).
The importance of PI3Kα in cancer is highlighted by the discovery that PIK3CA, the gene encoding the catalytic p110α subunit of PI3Kα, is frequently mutated in human malignancies.
In this case study, researchers sought insights into the oncogenic mechanism of two commonly expressed PI3Kα mutants by studying their conformational changes with molecular dynamics (MD) simulations, in comparison with the PI3Kα wild-type (normal, noncancerous) protein. The research team was particularly interested in evaluating the efficacy of cloud computing for performing MD simulations using GROMACS software. GRNET was used as the cloud computing service provider, providing a virtual machine with eight cores and GROMACS installation. Linear scaling was observed up to the eight available cores.
The team identified multiple benefits from the project:
- They gained an understanding of the cloud computing philosophy and what is involved in using a cloud-based solution for computational work.
- The team realized that cloud computing is an extremely easy process, as compared to building and maintaining your own cluster.
- They determined that moving computational work to the cloud during periods of full utilization of in-house compute resources is a viable approach to ensuring analysis throughput.
Open-Source Clinical Cancer Genomics Pipeline in the Cloud
Rapid progress in NGS is paving the way to individualized cancer treatment based on the sequencing of the cancer genome. This process is still evolving, and validated open source pipelines to process the enormous amounts of data involved are not available to everyone interested in this field.
Software programs used to align NGS data to the reference genome are governed by user-defined parameters and map the short-sequenced reads to the reference genome with varying accuracy. Most of the alignment tools require a long time to obtain accurate mappings of the short reads to the reference genome.
The authors of this case study aimed to start preparing guidelines on the optimal choices of open source alignment tools to be included into the NGS data-processing pipelines by matching these tools to the available computational resources. They had two primary objectives:
- Document how much time it took for Mapping and Assembly with Qualities (MAQ 0.6.6 and 0.7.1) and Short Read Mapping Package (SHRiMP 2.2.3) to align 2 million 75 base-pair (bp) length paired end sequence reads of cancer exome data to the human reference genome.
- Compute a complete mapping of the mast-cell leukemia exome data (tumor and germline available in Illumina Genome Analyzer FASTQ format) to the Human Genome Build 37 (GRCh37 hg19).
To enable portability of the computation environment (including the aligners, scripts, configuration settings) UberCloud Linux container images were used. The same image was used in the AWS, Bare Metal #1, and Bare Metal #2 test. The Linux container images were easily ported between environments by the Operations Engineer.
The team gained two particularly useful insights from the project:
- Some software programs are not built to take advantage of modern multiple-core architectures. A visualization approach was used to perform mapping with MAQ simultaneously on four compute nodes. Running simple programs in virtualization layers on HPC resources proved to be a very productive use of these resources, considerably reducing execution times for simple processes. Meanwhile, using UberCloud Linux containers on available HPC architecture made it possible to process big data much more efficiently.
- Alignment of the short reads to the reference genome in NGS exome analysis pipeline is only a single intermediate step in obtaining information on which genomic variants are present in the exome. The next step, search and identification of those genomic variants, is computationally demanding as well. It would be beneficial to assess a computational intensity and create resource usage guidelines for genomic variant identification tools such as Genome Analysis Toolkit (GATK), Samtools and MAQ.
 |
All three of these case studies are available in the UberCloud Compendium of Life Sciences Case Studies. The compendium includes nine case studies that explore innovative applications of cloud simulations in medicine and healthcare.
|

